Printable Version: Lab-Developed Tests and the FDA
Introduction
Advancements in personalized medicine have the potential to revolutionize health care. Genomics and molecular biology technologies are vital in the development of theranostics and other predictive and preventative areas of personalized medicine. However, few of these laboratory-developed-tests are currently regulated by the Food and Drug Administration (FDA) for analytical and clinical efficacy. Currently, the only oversight requirements governing LDTs are the laboratory requirements prescribed in the Clinical Laboratory Improvement Amendments of 1988 (CLIA).
While the FDA assumes authority for regulating LDTs, and is currently exercising enforcement discretion, it has indicated it will be increasing oversight of LDTs in the near future. FDA Commissioner Margaret A. Hamburg is now renewing FDA’s call for more active FDA regulation of LDTs and touting the Agency’s risk-based framework for regulating LDTs that is “under development.”
On July 9, 2012, President Obama signed into law the bipartisan FDA user-fee bill, the Food and Drug Administration Safety and Innovation Act (FDASIA). For the next five years, the Act prohibits the FDA from issuing guidance on LDT regulation unless the Agency provides a 60-day advance notice to the House Energy and Commerce Committee and the Senate Health, Education, Labor, and Pension Committee of its intent to take such action. This law all but places a time clock on the impending shift in the regulation of these tests.
Several stakeholders have submitted proposals to the FDA on risk-based strategies to facilitate tighter regulation on LDTs, among these are the College of American Pathologists (CAP) and the Advanced Medical Technology Association (AdvaMed). We have selected these proposals as highest likelihood of adoption.
Purpose
This article is a comparative study of the proposals by CAP and AdvaMed. It will analyze the proposed risk management strategies and comment on their likely impact on the clinical diagnostics industry should the FDA choose to accept their proposals. In addition, this article will propose a best-of-breed strategy to use as a template for any high-complexity CLIA laboratory in their assessment of the pending regulatory changes.
Scope
Only the proposals presented by the College of American Pathologists and AdvaMed as referenced below were considered when designing the best-of-breed strategy.
Analysis
The CAP Proposal Summary
The CAP proposes a risk-based model employing a public-private partnership to address oversight of LDTs. In their proposal, third-party accreditors and inspectors would oversee and monitor standards for low- and moderate-risk LDTs; high-risk LDTs would be reviewed directly by the FDA. We recognize that the CAP has a biased stake in these recommendations because they are the best-equipped accrediting agency to thrive from public/private partnership; however, it is a risk-balanced approach that also addresses the lack of currently-available resources by the FDA to regulate the LDT industry. The regulatory flexibility proposed by the CAP would encourage innovation of new diagnostic and predictive tests to promote and protect public health. Each laboratory would self-assess their LDT classification based on the FDA’s criteria for low-, moderate-, and high-risk tests. The determination would be verified by the laboratory’s certifier and/or accreditor (e.g. CAP, COLA, AABB, etc). Appendix A is a summary of the proposed tiers.
The proposal also states that there should be a harmonization between CMS’s CLIA standards and the FDA when it comes to quality systems management. Since the Quality Systems Management Standards of the FDA are more robust than those of CLIA, our opinion is that CLIA would most likely adopt the FDA standards, where applicable. In addition, the CAP proposes that certain direct-to-consumer tests which are currently unregulated would now fall under CLIA and have to follow the same guidance as other CLIA laboratories.
The AdvaMed Proposal Summary
Like the CAP proposal, AdvaMed suggests a harmonization of CLIA and FDA, and that all clinical laboratories should be subject to CLIA regulations. But unlike CAP, AdvaMed proposes that the FDA should oversee the safety and effectiveness of all diagnostic tests, whether that are made in a laboratory or by a manufacturer, because they all have the same risk/benefit profile for patients. Similarly to the CAP proposal, AdvaMed suggests a risk-based tiered approach for oversight focus. While well-standardized and low-risk tests could be exempted from and FDA premarket review, novel biomarkers using new technology could face the scrutiny of a Tier III FDA review. In between these two extremes, AdvaMed proposes using the existing three-tiered FDA definitions to categorize tests, where risk assessment and mitigation ability are used to further stratify classification. Appendix B is a summary of their approach.
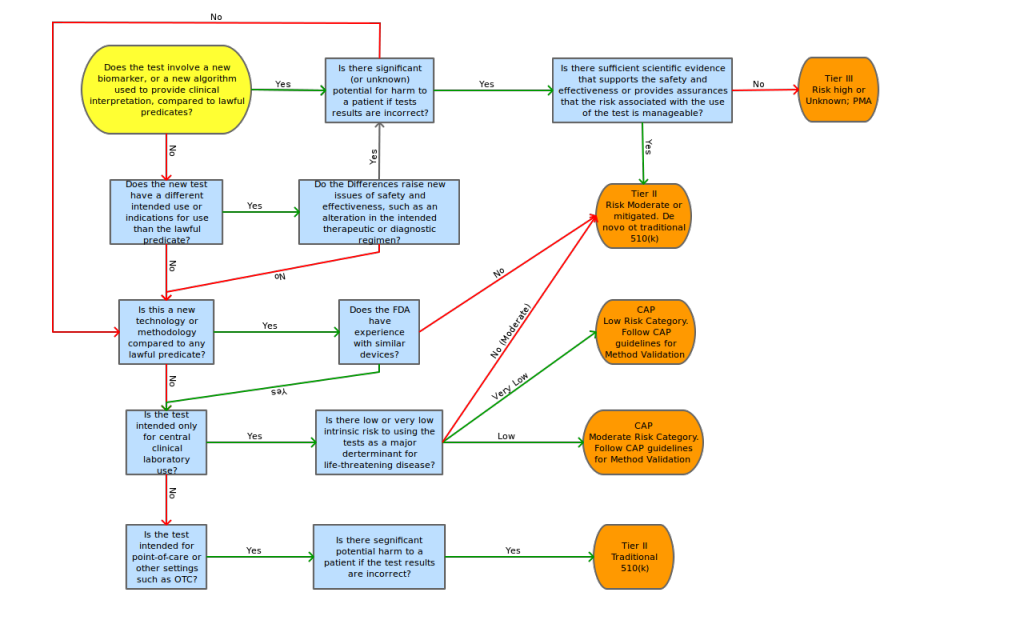
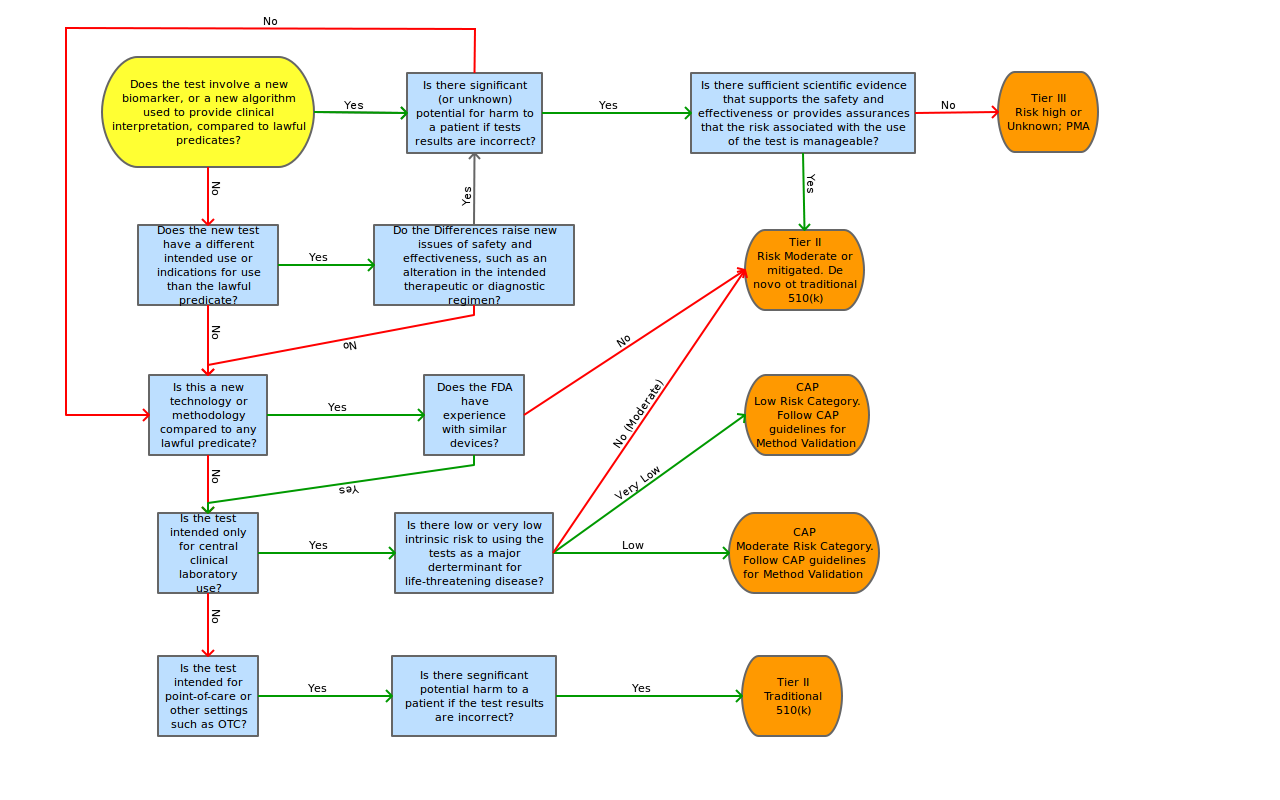
The AdvaMed proposal also contains a risk decision tree to aid in the determination of tier classification, risk assessment points of concentration and possible mitigating factors for these risk points.
Conclusion
While no one can accurately predict the political climate in the next 5 years, we can say that there is and will continue to be a significant push back from the nation’s largest clinical diagnostics companies for dramatic reforms in the CLIA/FDA regulatory areas such as those proposed by AdvaMed. It will cost these companies millions, maybe hundreds of millions of dollars to validate their systems to these new standards. It has the potential to stifle innovation and patient access to cutting-edge technology. Also, from a logistics point of view, the FDA does not currently have the resources to regulate the entire LDT industry.
The risk-based private/public partnership proposed by the CAP addresses both the logistics and the economic concerns of this issue while keeping patient safety in mind. Once considered high risk (as defined by the CAP proposal), it would then seem appropriate to classify an LDT as described in the AdvaMed proposal.
At Lab Insights, LLC, our current focus in the clinical diagnostics area is in the development of high-complexity, moderate to high risk tests run on established and new technologies. Many tests in our focus utilize multivariate algorithms, which disqualifies the tests from any FDA tier but Tiers II and III. It is most likely that when/if the FDA utilizes a risked-based approach to examine Laboratory-Developed Tests, the tests developed here will undergo a rigorous level of review.
To assess the level of regulation anticipated for FDA submission, we propose the adoption of a modified AdvaMed decision tree shown in Appendix C.
Under CAP, the following characteristics must be determined and approved by a Certified Lab Director for high-complexity testing as defined by CLIA (42 CFR 493.1443):
- Analytic Accuracy and Precision
- Analytic Sensitivity
- Analytic Specificity
- Analytic Interfering Substances
- Reportable Range
CAP Guidelines should be followed to comply with this regulation.
Also, under CAP, computer systems must be evaluated for:
- Computer Facility
- LIS/Computer Manual documentation
- Hardware and Software testing and documentation
- Training
- System Maintenance
- System Security
- Patient Result verification
References
- 2012. College of American Pathologists. 2012 CAP Checklists. http://www.cap.org
- 2010. College of American Pathologists. Proposed Approach to Oversight of Laboratory Developed Tests. http://www.cap.org
- 2012. Advanced Medical Technology Association. Risk-based Regulation of Diagnostics. www.advamed.org
- 2013. 42 CFR 493 – Laboratory Requirements. http://www.gpo.gov/fdsys/pkg/CFR-2003-title42-vol3/xml/CFR-2003-title42-vol3-part493.xml
- 2007. FDA Draft Guidance for Industry, Clinical Laboratories, and FDA Staff – In Vitro Diagnostic Multivariate Index Assays
NO EVENT SHALL LAB INSIGHTS, LLC BE LIABLE, WHETHER IN CONTRACT, TORT, WARRANT, OR UNDER ANY STATUTE OR ON ANY OTHER BASIS FOR SPECIAL, INCIDENTAL, INDIRECT, PUNITIVE, MULTIPLE OR CONSEQUENTIAL DAMAGES IN CONNECTION OR ARISING FROM LAB INSIGHTS, LLC SERVICES OR USE OF THIS DOCUMENT.
Appendix
Summary table of the AdvaMed Proposal Triage decision matrix
| Classification |
Determining Factors |
Oversight |
Low Risk:
the consequence of an incorrect result or incorrect interpretation is unlikely to lead to serious morbidity/mortality. |
The test result is typically used in conjunction with other clinical findings to establish or confirm diagnosis. No claim that the test result alone determines prognosis or direction of therapy. |
The laboratory internally performs analytical validation and determines adequacy of clinical validation prior to offering for clinical testing. The accreditor during the normally scheduled inspections will verify that the laboratory performed appropriate validation studies. |
Moderate Risk:
the consequence of an incorrect result or incorrect interpretation may lead to serious morbidity/mortality
AND the test methodology is well understood and independently verifiable. |
The test result is often used for predicting disease progression or identifying whether a patient is eligible for a specific therapy. The laboratory may make claims about clinical accuracy. |
The laboratory must submit validation studies to the CMS-deemed accreditor for review and the accreditor must make a determination that there is adequate evidence of analytical and clinical validity before the laboratory may offer the test clinically. |
High Risk:
the consequence of an incorrect result or incorrect interpretation cCaould lead to serious morbidity/mortality
AND the test methodology is not well understood or is not independently verifiable. |
The test is used to predict risk of, progression of, or patient eligibility for a specific therapy to treat a disease associated with significant morbidity or mortality, AND;
The test methodology uses proprietary algorithms or computations such that the test result cannot be tied to the methods used or inter-laboratory comparisons cannot be performed. |
The laboratory must submit test to FDA for review prior to offering the test clinically. CMS and accreditor determine compliance. |
Summary table of the AdvaMed Proposal Tiered decision matrix
| Category |
New (use of) Biomarker |
Established (use of) Biomarker |
| New Technology |
No Predicate devices (i.e. novel or high risk)
Little of no clinical literature
Requires analytical and clinical validation
Manufacturers and laboratories subject to premarket review
Tier III: PMA or de novo 510(k) |
Sufficient Clinical evidence to assess safety and effectiveness of biomarker
Requires analytical validation of new method on clinical specimens
Review level separated by FDA experience with technology
Tier II: traditional or de novo 510(k)
Tier I: traditional or streamlined 510(k), possible labeling review |
| Established Technology |
Could have predicate device
Little/no literature on biomarker, but literature and/or FDA experience with technology platform; moderate risk products
Manufacturers and laboratories subject to premarket review
Tier III: PMA or de novo 510(k)
Tier II: traditional or de novo 510(k) |
Sufficient Clinical evidence to assess safety and effectiveness of biomarker
Submission of labeling or data summarizing performance characteristics
Self certification/declaration of conformity with standards
Tier II: if moderate risk associated with use (traditional 510(k))
Tier I: if low risk associated with use (labeling review or streamlined 510(k))
Tier O: if risk low and managed, labeling review and/or consider exempt |
Proposed “Triage-then-Tier” decision tree by Lab Insights, LLC